

01.07.2021
Das Problem der Peakverbreiterung bei Multidetektorsystemen in der Gelpermationschromatographie
Dr. Gerhard Heinzmann, Alina Heinzmann, Sophia Heinzmann
In der Routineanalytik sind Gelpermeationschromatographiesysteme (GPC oder SEC) oft mit nur einem Detektor ausgestattet. Im Gegensatz zur HPLC, in der sich ein UV-Detektor als am häufigsten verwendeter Detektor etabliert hat, wird in der GPC meist ein Brechungsindexdetektor (RI = Refractive Index Detector) verwendet, da viele synthetische Polymere und Biopolymere keine chromophoren Gruppen besitzen und somit nicht oder nur sehr wenig UV-aktiv sind.
Leistungsfähige Gelpermeationschromatographiesysteme sind hingegen oft mit mehr als nur einem Detektor ausgestattet. Die bekannteste Detektorkombination ist sicherlich die Kombination eines RI-Detektors mit einem statischen Mehrwinkel-Lichtstreudetektor (MALS = Multi Angle Static Laser Light Scattering Detector). Mit einem solchen MALS/RI-System sind absolute Molekulargewichte und deren Verteilung zugänglich.
Oft wird noch ein Viskositätsdetektor hinzu geschaltet, der die Bestimmung von Verzweigungsstrukturen, auch von kleinen Polymermolekülen, ermöglicht. Falls doch mindestens ein Homopolymer des Copolymermoleküls UV-aktiv ist, kann als vierter Detektor ein UV-Detektor verwendet werden, der die Analyse der Zusammensetzungen von vielen Copolymermolekülen und Protein-Polysaccharid-Konjugaten ermöglicht.
Im Fall der Protein-Polysaccharid-Konjugate ist das Protein der UV-aktive Anteil des Konjugates. Ein Problem, das es bei Multidetektorsystemen immer zu beachten gilt, ist, dass jede Detektorzelle durch Ihr Volumen zu einer Verbreiterung des Analysenpeaks führt, ebenso wie die Verbindungskapillaren zwischen den Detektoren. Diese Peakverbreiterung muss im Rahmen der Kalibrierung eines Multidetektorsystems korrigiert werden, ebenso wie der Laufzeitunterschied zwischen den einzelnen Detektoren, das so genannte "Interdetector Volume".
Ursachen der Peakverbreiterung in Multidetektorsystemen
Wenn eine Probe nur ein einziges, klar definiertes, Molekulargewicht aufweist, wie dies z. B. bei einem Protein der Fall ist, dann würde man eigentlich einen sehr scharfen Probenpeak erwarten, ähnlich wie dies bei der Massenspektrometrie der Fall ist. In der Chromatographie führen aber eine ganze Reihe von physikalischen Effekten dazu, dass, anstelle eines scharfen Peaks, auch bei monodispersen Proben ein relativ breiter Peak resultiert.
Zunächst ist hier das interne Volumen des Chromatographiesystems zu nennen, maßgeblich das Volumen der Trennsäulen, aber auch das Volumen der Kapillaren. Der statistische Durchlauf der Probe durch die Trennsäulen führt zu einer deutlichen Peakverbreiterung. Diese Verbreiterung ist aber mathematisch sehr einfach zu korrigieren: man misst zunächst einen möglichst eng verteilten, oder im besten Fall sogar monodispersen, Standard; die Chromatographiesoftware kann dann, bei bekannter Polydispersität des Standards, die durch die Kapillaren und die Trennsäulen verursachte Peakverbreiterung korrigieren.
Komplexer wird die Mathematik, wenn man die zusätzliche Peakverbreiterung betrachtet, die durch die Detektoren im System entsteht. Die Detektoren eines Multidetektorsystems sind in aller Regel in Serie, also hintereinander, geschaltet. Nur beim Einsatz eines Vikositätsdetektors wird oft eine Parallelschaltung des Viskositätsdetektors mit dem RI-Detektor am Ende der Detektorreihenfolge bevorzugt.
All den genannten Detektorkombination ist gemein, dass der Probenpeak, der in den einzelnen Detektoren erzeugt wird, immer breiter wird, je weiter hinten sich der Detektor in der Detektionsreihenfolge befindet. Diese Peakverbreiterung wird durch zwei wesentliche Dinge verursacht:
- die Kapillaren, die die einzelnen Detektoren verbinden.
Jede Kapillare besitzt ein gewisses internes Volumen, das zur Verbreiterung des Probenpeaks beiträgt. Im Englischen wird dieses Kapillarvolumen zwischen den Detektoren als "Interdetector Volume" bezeichnet. - die Messzellen der einzelnen Detektoren.
Das interne Volumen der Messzellen in den Detektoren führt ebenfalls zur Verbreiterung des Probenpeaks.
Während das interne Volumen der Messzellen des UV-Detektors und auch des MALS-Detektors eher gering ist, kommt dem Viskositätsdetektors und auch dem RI-Detektor eine Sonderrolle zu. Der Viskositätsdetektor hat ein vergleichsweise großes internes Volumen, da die Messzelle hier ein Kapillarsystem mit einer oder mehreren Verzögerungskolonnen darstellt. Der RI-Detektor hat ebenfalls ein großes internes Volumen, da sich im Auslass des Detektors ein Ventil zur Spülung der Referenzzelle befindet, und die Auslasskapillare in der Regel ein großes inneres Volumen aufweist, um den Rückdruck auf die Messzelle des RI-Detektors möglichst gering zu halten.
Das Problem der systembedingten Peakverbreiterung
Ein breiterer Peak steht messtechnisch eigentlich für eine größere Polydispersität der Probe. Wird die Peakverbreiterung aber nur durch das Messsystem, und nicht durch die Probe selbst erzeugt, dann ist die scheinbar resultierende Polydispersität der Probe natürlich zu groß. Daher muss man versuchen, die Peakverbreiterung, die durch das Messsystem verursacht wird, mathematisch zu korrigieren und somit, möglichst weitestgehend, zu eliminieren.
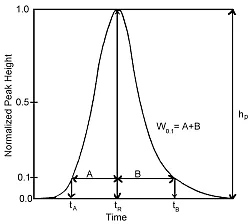
- Abb.1 :Mathematische Korrektur der
systembedingten Peakverbreiterung [1]
Lösungswege zur Korrektur der systembedingten Peakverbreiterung
Eine Möglichkeit, die systembedingte Peakverbreiterung zu korrigieren, ist, dass man das System mit einem eng verteilten Polymer- oder Biopolymerstandard kalibriert. Idealerweise kennt man die Polydispserität des Kalibrierstandards möglichst genau und kann diese in der Software auch angeben.
Bei der Kalibrierung kann dann die von Detektor zu Detektor zunehmende Peakverbreiterung berücksichtigt und mathematisch korrigiert werden. Der Probenpeak kann z. B. bei 10% Peakhöhe in der Peakmitte betrachtet, und die Verbreiterung in beide Richtungen des Elutionsvolumens bzw. der Retentionszeit berücksichtigt werden (Abbildung 1).
Die Peakverbreiterung kann mathematisch nun durch zwei Parameter, Sigma (σ) und Tau (τ), ausgedrückt werden:
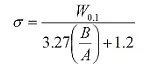
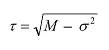
Wobei gilt: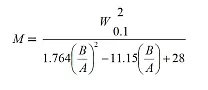
Nicht systembedingte Peakverbreiterung
Die Probenpeaks der einzelnen Detektoren werden sich allerdings in der Regel niemals perfekt überlagern, da neben der systembedingten Peakverbreiterung auch noch weitere probenbedingte Peakverbreiterungen existieren. Ein einfaches Beispiel soll anhand der Kombination eines Brechungsindex-Detektors mit einem statischen Mehrwinkel-Lichtstreudetektor beschrieben werden.
Hier kann es bei den beiden Detektorpeaks bei einer breit verteilten Probe sowohl am hochmolekularen Ende als auch am niedermolekularen Ende zu Unterschieden in der Peakform kommen, die alleine von der Probe herrühren und keineswegs systembedingt sind.
Betrachtet man zunächst das hochmolekulare Ende des Peaks, so kann es sein, dass die Probe sehr geringe Mengen an sehr hochmolekularem, ggf. verzweigtem, Material enthält. Diese geringe Menge kann zu wenig sein, als dass der nicht sehr empfindliche Brechungsindexdetektor dies erkennen könnte. Durch das sehr hohe Molekulargewicht dieses Probenanteils wird sich aber im Mehrwinkel-Lichtstreudetektor ein deutlicher Peak abzeichnen.
Ähnliches gilt in umgekehrter Reihenfolge auch für den niedermolekularen Bereich des Probenpeaks. Hier können noch wesentliche niedermolekulare Probenanteile im Brechungsindexdetektor sichtbar sein, während der Mehrwinkel-Lichtstreudetektor bereits wieder zu seiner Basislinie zurückgekehrt ist, da die Molekulargewichte dieser Probenbestandteile zu gering sind, als dass sie ein Signal im molekulargewichtssensitiven Mehrwinkel-Lichtstreudetektor erzeugen würden.
Applikationsbeispiel aus der Gelpermeationschromatographie
Abbildung 2 zeigt die Rohdaten einer Messung eines eng verteilten Polymerstandards mit einem Molekulargewicht von 100.000 g/mol mit der GPC/SEC. Als Detektoren wurden in Reihe geschaltet nacheinander ein MALS-Detektor (blau) und ein RI-Detektor (rot) verwendet.
Es ist deutlich zu erkennen, dass die Peaks der einzelnen Detektoren zeitlich versetzt eluieren, und zwar in der Reihenfolge, in der die Detektoren durchlaufen werden. Außerdem ist der Peak des RI-Detektors etwas breiter als der Peak des MALS-Detektors, da die Messzelle des MALS-Detektors zu einer zusätzlichen Peakverbreiterung führt, die im RI-Detektor sichtbar wird.
Abbildung 3 zeigt dieselbe Messung nach der Kalibrierung und Korrektur der Peakverbreiterung sowie der Laufzeitunterschiede. Beide Peaks überlagern sich nun nahezu perfekt.
Abbildung 4 zeigt die Messung einer breit verteilten Polymerprobe mit einem Molekulargewicht von 250.000 g/mol und einer Polydispersität von 2,5 (Mw/Mn).
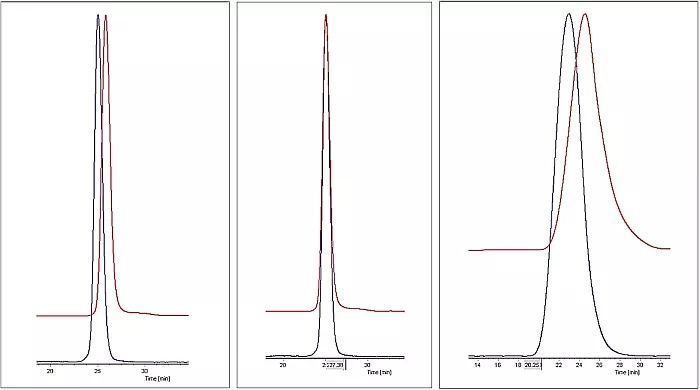
v.l.n.r.: Abb.2: MALS (blau) und RI (rot) Rohdaten
Abb 3: MALS- (blau) und RI-Signal (rot) überlagert nach Kalibration und Korrektur der Laufzeitunterschiede und der σ- und τ-Werte
Abb.4: Breit verteilter Polymerstandard nach Kalibration und Korrektur der Laufzeitunterschiede und der σ- und τ-Werte MALS-Signal (blau) und RI-Signal (rot)
Der RI-Detektor spricht auf die Konzentration der Probe an, während der MALS-Detektor primär auf das Molekulargewicht der Probe anspricht. Das Maximum der Probenkonzentration und das Maximum des Molekulargewichtes der Probe liegen aber nicht an derselben Stelle des Elutionsvolumen, daher überlagern sich die Peaks der beiden Detektoren trotz Korrektur der Peakverbreiterung und der Laufzeitunterschiede nicht.
Fazit
Werden in einem GPC-System mehrere Detektoren miteinander kombiniert, dann ist es sehr wichtig, dass die resultierende Peakverbreiterung der einzelnen Detektoren sowie der Laufzeitunterschied zwischen den Detektoren im Rahmen der Systemkalibrierung mathematisch erfasst und korrigiert bzw. eliminiert wird. Dies kann mittels eines eng verteilten Polymerstandards durchgeführt werden.
Allerdings muss bei der Peakverbreiterung zwischen einer systembedingten Peakverbreiterung und einer probenbedingten Peakverbreiterungen unterschieden werden. Während die systembedingte Peakverbreiterung zu falschen Resultaten in der Polydispersität einer Probe führen würde, und daher korrigiert werden muss, ist die probenbedingte Peakverbreiterung eine wirklich vorhandene Eigenschaft der Probe. Eine Korrektur ist in diesem Fall nicht notwendig, sie würde, ganz im Gegenteil, die Resultate verfälschen.
Wichtig ist auch zu wissen, dass sich die Detektorpeaks einer breit verteilten Probe trotz Korrektur der Peakverbreiterung und der Laufzeitunterschiede nicht überlagern, da die einzelnen Detektoren auf unterschiedliche physikalische Eigenschaften der Probe ansprechen.
Literatur
[1] Viscotek Corporation, "GPC/SEC Masterclass", Houston, Texas, USA, 1999